Research method finds new use in diagnosis of genetic disorders
This new testing method represents a paradigm shift in laboratory genetics, moving from the traditional DNA-centric approach to one focusing on the patient’s cells.
Read More

This new testing method represents a paradigm shift in laboratory genetics, moving from the traditional DNA-centric approach to one focusing on the patient’s cells.
Read More
The researchers hope that in the future their findings could be used as a marker of effective therapeutic response.
Read More
The results support germline testing for cancer-predisposition variants among children with rhabdomyosarcoma, which could aid in early clinical surveillance strategies.
Read More
Researchers visualize in atomic detail the first step of the mechanism that prompts DNA gyrase for resolving DNA entanglements.
Read More
This study opens new avenues of research in human and genome biology and eventually will help to bring potential new treatments to patients and their families.
Read More
After 64 schools competed and about a quarter million votes were cast, Baylor College of Medicine took the win home!
Read More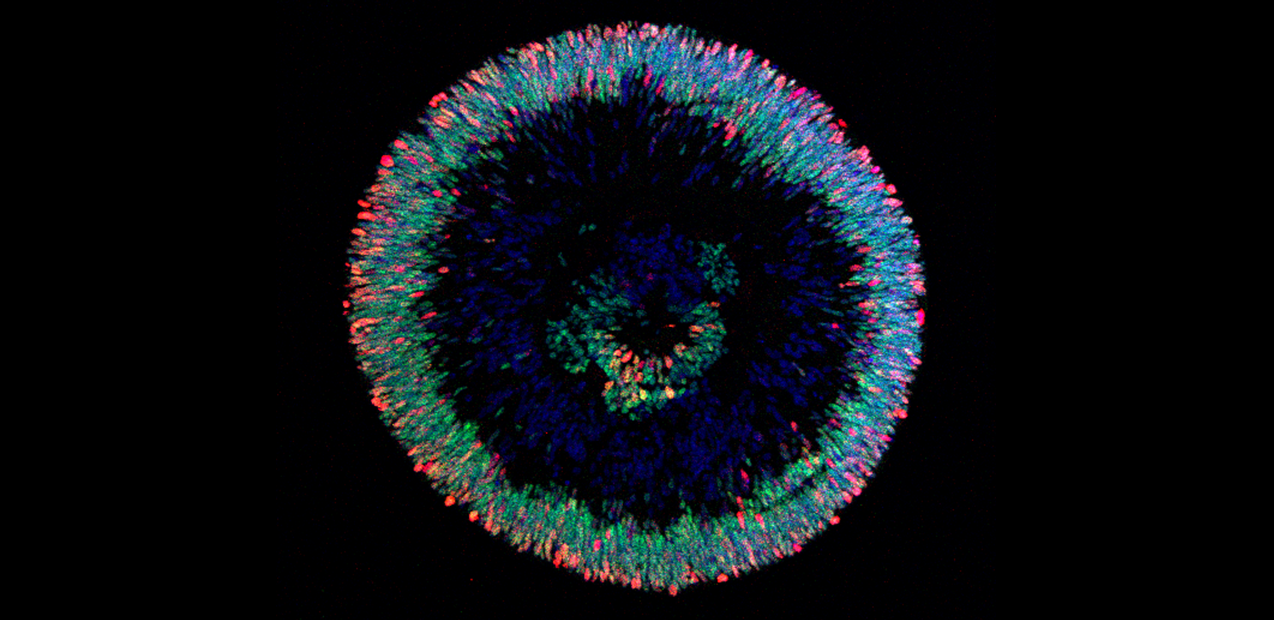
Retinal organoids are helping researchers study human retinal development and eye diseases such as glaucoma and macular degeneration.
Read More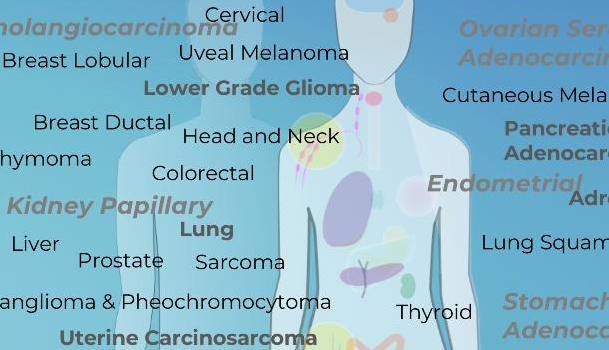
The genes with cancer-relevant associations arising in this study would represent strong candidates for further investigation on their value in genetic testing.
Read More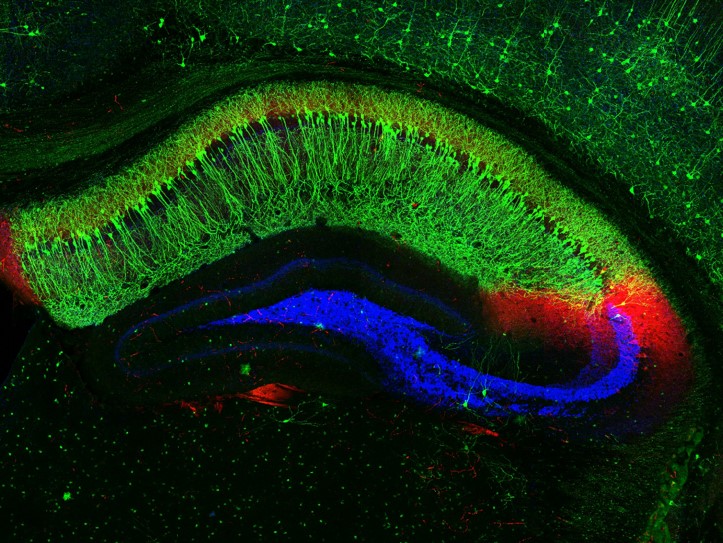
Researchers show for the first time the role endocannabinoid signals play in living animals moving about in the environment.
Read More
Learn about the wastewater epidemiology project that has advanced to the final stage of the competition and vote!
Read More